De la mà de la investigadora Sonia Guil coneixem la darrera hora quant a tractament de la malaltia amb un prometedor medicament aprovat per la FDA americana
Mes: maig de 2023

Quin és el tractament?
Fins ara no existeix un tractament farmacològic per evitar, eliminar o millorar els símptomes de la Síndrome de Rett, però si que existeix medicació per controlar les convulsions i altres teràpies que poden millorar significativament la qualitat de vida de les nenes. Els medicaments que s’han utilitzat o estudiat són: la Bromocriptina, el Dextrometorfano, el Folato i betaína. La L-carnitina també pot ajudar a millorar les habilitats del llenguatge, la massa muscular, la lucidesa mental, igual que l’energia i la qualitat de vida, mentre disminueix el restrenyiment i el somni durant el dia. LaL-dopa pot reduir la rigidesa motriu en les etapes posteriors de la malaltia. Per descomptat, qualsevol d’aquests medicaments ha de ser prescrit per un metge.
El tractament s’ha de fer amb l’acompanyament d’un equip multidisciplinari, incloent-hi un neuròleg, fisioterapeuta, terapeuta ocupacional, fonoaudiòleg i nutricionista, i inclou:
Fisioteràpia i hidroteràpia, que s’han de fer per millorar la força muscular, la postura i la respiració, a més d’alleujar l’escoliosi i millorar l’equilibri i la marxa.
Teràpia ocupacional, per ajudar a reduir els moviments repetitius i recuperar els moviments de les mans, millorant les activitats diàries, com menjar i vestir-se;
Fonoaudiologia, per ajudar la parla i i en el llenguatge no verbal, millorant la comunicació i interacció social;
Nutrició, que promou una alimentació balancejada i adequada, ajudant a mantenir un desenvolupament adequat, a més d’evitar vòmits, reflux, restrenyiment, ennuegaments i pèrdua de pes.
Es recomana la fisioteràpia intensa al més aviat possible per prevenir escoliosi, rigidesa, peu equí o unes altres i afavorir alhora la mobilitat. Així mateix, existeixen una sèrie d’aparells ortopèdics que poden evitar els “peus de ballarina”, les mans juntes o altres patologies. Han de ser recomanats per fisioterapeutes i metges després de valorar cada cas.
També la teràpia musical ha estat utilitzada a Europa des de 1972 amb èxit i és considerada com un mitjà per comunicar-se.
Es creu que és beneficiós el reduir les estereotípies manuals i augmentar l’atenció de la nena i la cerca de contacte visual. És important afavorir la marxa i tot moviment voluntari, així com insistir sobre la importància d’exercicis de reeducació funcional, que permetin limitar les deformacions i mantenir al màxim possible la independència. És necessari parar esment a la higiene bucal i ser regular en tots els tractaments que puguin ajudar a millorar el present i el futur de les nenes.
Un percentatge important de les noies i dones amb la síndrome de Rett poden patir epilèpsia, per la qual cosa és convenient tenir un seguiment realitzat pel neuro-pediatre.
Símptomes
Per poder diagnosticar la síndrome de Rett (anomenat també trastorn de Rett) cal complir dos criteris. Aquests fan referència als seus símptomes:
Criteri A: calen tres condicions:
Desenvolupament pre i peri natal normals.
Desenvolupament psicomotor normal fins als 5 mesos.
Circumferència cranial normal al naixement.
Criteri B: entre els 5 i els 48 mesos s’ha de complir el següent:
Desacceleració del creixement cranial.
Pèrdua d’habilitats manuals adquirides (entre els 5 i els 30 mesos de vida).
Pèrdua de la implicació social.
Atàxia de la marxa.
Greu alteració del llenguatge i de làrea psicomotora.
Els símptomes poden ser variables i d’intensitat diversa:
- Problemes respiratoris que es poden accentuar per situacions d’estrès i que es presenten quan la nena està desperta, no mentre dorm.
- Canvis en el desenvolupament físic i intel·lectual.
- Problemes motors com a dificultat en caminar i moviments repetitius de les mans.
- Trastorns gàstrics com a restrenyiment o reflux gastroesofàgic.
- Problemes circulatoris i extremitats flàccides.
- Variació dels patrons normals de somni.
Descoberta la implicació del genoma Fosc
El síndrome de Rett és una malaltia del desenvolupament neurològic que constitueix la segona causa més freqüent de retard mental en dones, després de la Síndrome de Down.
El quadre clínic comença a aparèixer 6-18 mesos després del naixement i consisteix en una pèrdua de capacitats cognitives, socials i motores acompanyada de comportaments autístics com, per exemple, moviments esterotipats de les mans. Avui dia no existeix tractament efectiu de la malaltia més enllà del control
de la seva simptomatologia.
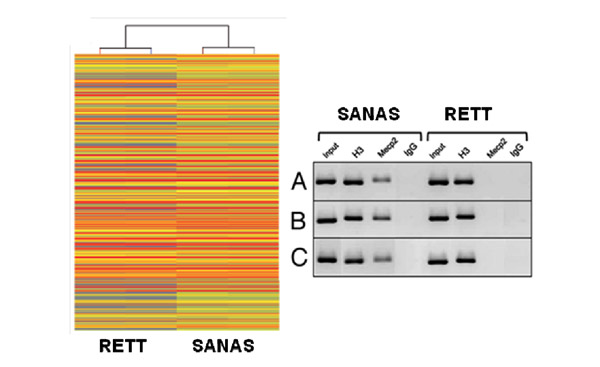
La síndrome sol ser a causa de la presència d’una mutació al gen MeCP2, un gen epigenètic que com si fos un imant regula l’expressió de molts altres gens de la cèl·lula. No obstant això, encara hi ha un gran desconeixement de les alteracions moleculars presents a la malaltia. El grup del Dr Manel Esteller, Director del Programa d’Epigenètica i Biologia del Càncer de l’Institut de Investigacions Biomedicas de Bellvitge, Investigador ICREA i Professor de Genètica de la Universitat de Barcelona, descriu aquesta setmana a la revista biomèdica internacional RNA Biology la identificació de fils especials de àcid ribonucleic (ARN) alterades a la Síndrome de Rett denominades ARNs no codificants de llarga cadena (lncARNs). Els lncARNs actuarien com agents supervisors encarregats d’apagar o encendre altres gens de el nostre genoma i així regular l’activitat de les neurones.
Els investigadors van recórrer a un model de raó que reproduïa realment les característiques de la síndrome de Rett humà i en el mateix compararon l’expressió de les cadenes llargues de ARN en animals sans y malalts. El grup liderat pel Dr Manel Esteller ha descobert que la presencia de mutacions en el gen Mecp2 provocava alteracions en la activitat dels ARN llargs no codificats originats a partir del “genoma oscuro”, aquell on no es troben els gens típics ni produeixen proteïnes.
Entre els lncARNS identificats com a alterats a les regions cerebrals afectades en la síndrome de Rett es troba un que regula la funció del receptor GABA. Esta ultima molecula (cuyas sigles es refereixen a l’àcid gamma- aminobutírico) és un neurotransmisor clau en el sistema nervioso cerebral de tots els vertebrats i la seva alteració podrien explicar els defectes de comunicació neurona-neurona a les nenes afectades per la síndrome de Rett. Aquests descobriments, a més d’augmentar el coneixement sobre les causes d’aquesta malaltia, podria tenir conseqüències futures per al tractament d’aquests pacients ja que teràpies que tuvieran com diana els lncARNs o el receptor GABA mereix ser estudiades en assaigs preclínics.
L’estudi ha estat recolzat pel Departament de Salut de la Generalitat de Catalunya, la Institució Catalana d’Estudis Avançats (ICREA), el Ministerio
de Sanidad y Consumo (E-RARE), los Proyecto Europeos DISCHROM y EPINORC, la Fondation Lejeune (Francia) i l’Associació Catalana del Síndrome de Rett.
Article: Desregulació del transcriptoma llarg d’ARN no codificant en un Rett model de ratolí de síndrome. Petazzi P, Sandoval J, Szczesna K, Jorge OC, Roa L,
Sayols S, Gomez A, Huertas D, Esteller M. RNA Biology, 10(7), 2013.

Descripcio de la malaltia
La síndrome afecta gairebé exclusivament a nenes i dones. Això es deu al fet que està principalment causat per una mutació esporàdica en el gen MECP2 del cromosoma X. La funció d’aquest gen és regular el funcionament d’altres gens, per la qual cosa juga un paper fonamental en el desenvolupament cerebral.
A Espanya, unes 2.700 nenes pateixen l’anomenada Síndrome de Rett, una malaltia neurològica que afecta la mobilitat i que causa un autisme sever.
La síndrome de Rett és un trastorn neurològic infantil de base genètica caracteritzat per una evolució normal inicial seguida de la pèrdua de l’ús voluntari de les mans, un creixement retardat del cervell i del cap, dificultats per caminar, convulsions i retard mental.
Els nens que pateixen la síndrome de Rett generalment comencen a perdre la capacitat de parlar, fer contacte visual i comunicar-se d’altres maneres. Poden perdre l’interès en les altres persones, joguines i entorn. Alguns nens pateixen els canvis, com la pèrdua sobtada de la parla, ràpidament
Alteracions en altres gens com el CDLK5 i el FOXG1 també produeixen síndrome de Rett, encara que els símptomes i l’afectació poden ser molt diferents.
Aquesta síndrome es diagnostica observant signes i símptomes durant el creixement inicial i el desenvolupament de la nena i realitzant avaluacions periòdiques del seu estat físic i neurològic.
Hi ha nombrosos problemes crònics de salut i comportament associats amb RTT per als quals seria raonable esperar dolor o malestar (p. ex., escoliosi, restrenyiment i problemes gastrointestinals relacionats, comportament auto agressiu)
Existeix també una prova genètica per confirmar el diagnòstic clínic d’aquest trastorn. La prova involucra buscar la mutació de tipus MECP2 en el cromosoma X del nen o nena i pot identificar fins a un 80% dels casos.
La síndrome de Rett és una malaltia de les anomenades rares, la qual cosa significa que afecta menys de cinc casos per cada 10.000 persones. Rett és un trastorn greu del neurodesenvolupament d’origen genètic que afecta de manera gairebé exclusiva al sexe femení i que porta a una discapacitat greu. També afecta gairebé tots els aspectes de la vida d’una persona ja que impedeix la seva capacitat per parlar, caminar, menjar i fins i tot respirar de forma normal.
Malgrat les dificultats que impliquen els símptomes, la majoria dels individus que pateixen de la Síndrome de Rett continúen vivint bé fins a l’edat adulta o major.
El 1966, un neuròleg austríac anomenat Andreas Rett va descriure per primera vegada més de 20 joves pacients de sexe femení que compartien característiques similars, a partir de l’observació d’idèntics moviments estereotípics de les mans.
El diagnòstic del SR es basa en criteris clínics, però els avenços en la biologia molecular i en la genètica en particular han obert el ventall de possibilitats diagnòstiques a les diferents formes clíniques que abans quedaven sense classificar, alhora que l’anàlisi molecular permet confirmar el criteri clínic i aportar informació quant al pronòstic del pacient.