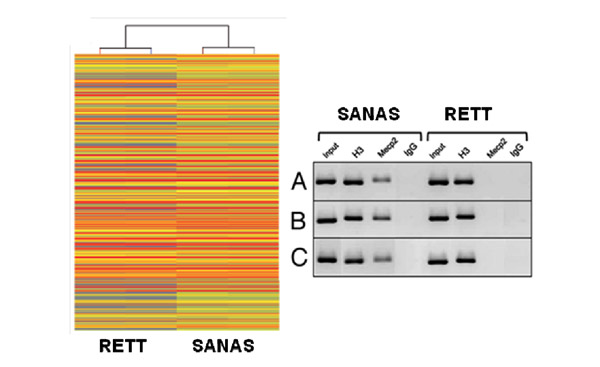
La síndrome de Rett, un trastorn neurològic d’origen genètic que afecta principalment nenes, ha estat objecte d’estudi durant dècades a causa del seu impacte devastador en el desenvolupament cognitiu, motor i social. Aquesta condició, causada per mutacions al gen MECP2, continua sent un desafiament per a la comunitat científica, però una investigació recent en què participa la Universitat de València ha fet un pas significatiu cap a la comprensió i el maneig d’aquesta malaltia. Segons una notícia publicada al portal de la Facultat de Ciències Biològiques de la UV, un equip d’investigadors ha aconseguit resultats que podrien endarrerir l’avenç de la síndrome de Rett obrint noves esperances per a pacients i famílies.