Gràcies a l’associació espanyola de Sindrome de Rett compartim amb vosaltres aquest interessant vídeo sobre un recent estudi i avaluació expressió del dolor
Informació en català sobre Rett. No som cap organisme oficial. Amics i familiars afectats per aquesta malaltia.
Gràcies a l’associació espanyola de Sindrome de Rett compartim amb vosaltres aquest interessant vídeo sobre un recent estudi i avaluació expressió del dolor

Parlar d’aquesta síndrome és fer-lo d’una malaltia rara, i que té el seu origen en una mutació genètica. La malaltia de Rett afecta al neurodesenvolupament, arribant a alterar el comportament d’altres gens i tenint la seva principal expressió, l’impediment d’algunes funcions motores i epilèpsia. En aquest article t’explicarem amb més detall què és la síndrome de Rett i quina és la seva afectació en la vida afectiva i sexual de les persones malaltes.
Durant l’espermatogènesi, es donen lloc a una sèrie d’alteracions de gens i provocant una sèrie de problemes que, de manera més freqüent afecta a les nenes.
Això es deu al fet que el gen que ha mutat es troba en un cromosoma X, i quan l’organisme detecta el problema, aquest és substituït per un altre en existir una còpia, però els individus homes no compten amb aquest cromosoma que serveix de substitut, per tant, es produeix una mort perinatal o abans del naixement. En tot cas, existeix una variant d’aquesta síndrome de Rett amb símptomes similars i que sí que es dona en els nens.
Per descomptat, en tractar-se d’una malaltia rara, les fonts de finançament provenen d’àmbits privats, amb el que els avanços són massa lents. És el principal problema amb el qual es troben els familiars de persones afectades per la síndrome de Rett. Aconseguir finançament, sempre és bastant complicat, i qualsevol iniciativa d’investigar de manera pública compten amb nombroses traves administratives que, en la major part dels casos, impedeix continuar amb les recerques.
Els símptomes d’aquesta malaltia triguen a aparèixer, perquè les nenes neixen sanes en aparença i res fa sospitar d’aquests problemes. És aproximadament a partir dels sis mesos quan els pares perceben que comença a perdre habilitats. Per exemple, si ja ha començat a fer els seus primers passos, deixa de fer-ho, apareixen moviments repetitius a les mans. També és molt habitual que fugin de la interacció social, fins i tot amb els pares. No es deixen abraçar o besar amb facilitat. Però un dels problemes més evidents és que la síndrome de Rett porta associada una varietat d’epilèpsia, que no pot ser tractada amb fàrmacs, la qual cosa dificulta la vida de les persones, malaltes i dels seus cuidadors.
La síndrome de Rett compta amb diferents fases, els científics han catalogat quatre d’elles. L’inici prematur, la destrucció accelerada, l’estabilització i la deterioració motora tardana.
Aquesta malaltia condiciona la vida de les persones que la pateixen, òbviament també aquelles que estan al seu càrrec, encara que no existeixen els mateixos graus d’afectació. Els símptomes que es donen en tots els malalts tenen relació amb la capacitat motora, com poder agarrar objectes. També compten amb dificultats en la parla, problemes d’equilibri i coordinació o discapacitats intel·lectuals.
D’altra banda, la respiració també sofreix dificultats en forma d’hiperventilació, la qual cosa al costat dels problemes socials i de conductes fa d’ella una malaltia que requereix vigilància permanent.
S’està investigant sobre la manera de poder millorar la qualitat de vida i s’ha descobert un derivat del cànnabis que pot tenir efectes positius sobre aquests símptomes.
De la mateixa manera, el tractament de fisioteràpia es perfila com una manera de mantenir el to muscular i no continuar perdent capacitats. Per a aquests malalts, recuperar les habilitats socials és fonamental, per la qual cosa un tractament que té molta efectivitat és la equinoteràpia, ja que aconsegueixen establir un vincle molt sòlid en tot moment amb aquests animals.
La vida sexual d’aquestes persones compta amb evidents dificultats. D’una banda, per la falta de relació amb uns altres, ja que sofreixen una deterioració molt greu en les seves relacions socials i conductuals.
De la mateixa manera, la síndrome de Rett porta aparellat una gran quantitat de problemes motors que, en molts casos, porta als malalts a romandre prostrat en llit o una cadira de rodes, per molt de temps. Per tant, existeix una dificultat màxima a l’hora d’establir vincles amb altres persones, en estar les seves vides molt limitades.
Però això no vol dir que un malalt de Rett no compti amb les necessitats de satisfacció pròpies de qualsevol individu. Per tant, l’autoconeixement del cos i l’autosatisfacció són les principals maneres en les quals un malalt d’aquestes característiques pot gaudir de la sexualitat. És recomanable buscar estímuls sexuals per exemple a internet per ajudar a millorar aquesta vida sexual. Webs de vídeos porno per a adults com pornogratisdiario.com són molt valorades. I és que la sexualitat per internet ajuda molt persones amb alguna dificultat per establir relacions socials. Llocs web experts en lleure per a adults com a videospornogratisx.net poden servir d’ajuda tal com hem indicat.
Per tot això, els afectats per la síndrome de Rett no solament han d’enfrontar-se a les traves pròpies d’una malaltia rara que els incapacita, sinó a la dificultat, afegida de tenir una vida social que no és del tot plena i que acaba llastrant la resta d’aspectes de la seva vida.
De la mà de la investigadora Sonia Guil coneixem la darrera hora quant a tractament de la malaltia amb un prometedor medicament aprovat per la FDA americana

Fins ara no existeix un tractament farmacològic per evitar, eliminar o millorar els símptomes de la Síndrome de Rett, però si que existeix medicació per controlar les convulsions i altres teràpies que poden millorar significativament la qualitat de vida de les nenes. Els medicaments que s’han utilitzat o estudiat són: la Bromocriptina, el Dextrometorfano, el Folato i betaína. La L-carnitina també pot ajudar a millorar les habilitats del llenguatge, la massa muscular, la lucidesa mental, igual que l’energia i la qualitat de vida, mentre disminueix el restrenyiment i el somni durant el dia. LaL-dopa pot reduir la rigidesa motriu en les etapes posteriors de la malaltia. Per descomptat, qualsevol d’aquests medicaments ha de ser prescrit per un metge.
El tractament s’ha de fer amb l’acompanyament d’un equip multidisciplinari, incloent-hi un neuròleg, fisioterapeuta, terapeuta ocupacional, fonoaudiòleg i nutricionista, i inclou:
Fisioteràpia i hidroteràpia, que s’han de fer per millorar la força muscular, la postura i la respiració, a més d’alleujar l’escoliosi i millorar l’equilibri i la marxa.
Teràpia ocupacional, per ajudar a reduir els moviments repetitius i recuperar els moviments de les mans, millorant les activitats diàries, com menjar i vestir-se;
Fonoaudiologia, per ajudar la parla i i en el llenguatge no verbal, millorant la comunicació i interacció social;
Nutrició, que promou una alimentació balancejada i adequada, ajudant a mantenir un desenvolupament adequat, a més d’evitar vòmits, reflux, restrenyiment, ennuegaments i pèrdua de pes.
Es recomana la fisioteràpia intensa al més aviat possible per prevenir escoliosi, rigidesa, peu equí o unes altres i afavorir alhora la mobilitat. Així mateix, existeixen una sèrie d’aparells ortopèdics que poden evitar els “peus de ballarina”, les mans juntes o altres patologies. Han de ser recomanats per fisioterapeutes i metges després de valorar cada cas.
També la teràpia musical ha estat utilitzada a Europa des de 1972 amb èxit i és considerada com un mitjà per comunicar-se.
Es creu que és beneficiós el reduir les estereotípies manuals i augmentar l’atenció de la nena i la cerca de contacte visual. És important afavorir la marxa i tot moviment voluntari, així com insistir sobre la importància d’exercicis de reeducació funcional, que permetin limitar les deformacions i mantenir al màxim possible la independència. És necessari parar esment a la higiene bucal i ser regular en tots els tractaments que puguin ajudar a millorar el present i el futur de les nenes.
Un percentatge important de les noies i dones amb la síndrome de Rett poden patir epilèpsia, per la qual cosa és convenient tenir un seguiment realitzat pel neuro-pediatre.
Per poder diagnosticar la síndrome de Rett (anomenat també trastorn de Rett) cal complir dos criteris. Aquests fan referència als seus símptomes:
Criteri A: calen tres condicions:
Desenvolupament pre i peri natal normals.
Desenvolupament psicomotor normal fins als 5 mesos.
Circumferència cranial normal al naixement.
Criteri B: entre els 5 i els 48 mesos s’ha de complir el següent:
Desacceleració del creixement cranial.
Pèrdua d’habilitats manuals adquirides (entre els 5 i els 30 mesos de vida).
Pèrdua de la implicació social.
Atàxia de la marxa.
Greu alteració del llenguatge i de làrea psicomotora.
Els símptomes poden ser variables i d’intensitat diversa:
El síndrome de Rett és una malaltia del desenvolupament neurològic que constitueix la segona causa més freqüent de retard mental en dones, després de la Síndrome de Down.
El quadre clínic comença a aparèixer 6-18 mesos després del naixement i consisteix en una pèrdua de capacitats cognitives, socials i motores acompanyada de comportaments autístics com, per exemple, moviments esterotipats de les mans. Avui dia no existeix tractament efectiu de la malaltia més enllà del control
de la seva simptomatologia.
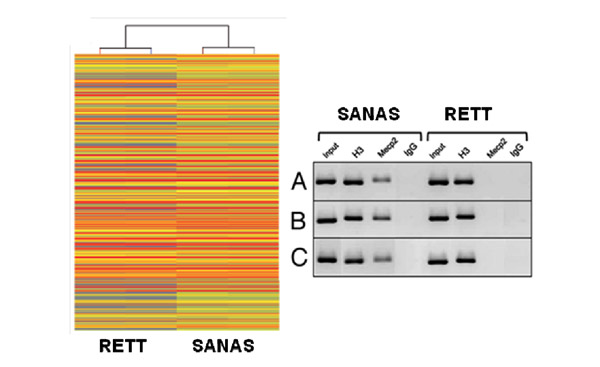
La síndrome sol ser a causa de la presència d’una mutació al gen MeCP2, un gen epigenètic que com si fos un imant regula l’expressió de molts altres gens de la cèl·lula. No obstant això, encara hi ha un gran desconeixement de les alteracions moleculars presents a la malaltia. El grup del Dr Manel Esteller, Director del Programa d’Epigenètica i Biologia del Càncer de l’Institut de Investigacions Biomedicas de Bellvitge, Investigador ICREA i Professor de Genètica de la Universitat de Barcelona, descriu aquesta setmana a la revista biomèdica internacional RNA Biology la identificació de fils especials de àcid ribonucleic (ARN) alterades a la Síndrome de Rett denominades ARNs no codificants de llarga cadena (lncARNs). Els lncARNs actuarien com agents supervisors encarregats d’apagar o encendre altres gens de el nostre genoma i així regular l’activitat de les neurones.
Els investigadors van recórrer a un model de raó que reproduïa realment les característiques de la síndrome de Rett humà i en el mateix compararon l’expressió de les cadenes llargues de ARN en animals sans y malalts. El grup liderat pel Dr Manel Esteller ha descobert que la presencia de mutacions en el gen Mecp2 provocava alteracions en la activitat dels ARN llargs no codificats originats a partir del “genoma oscuro”, aquell on no es troben els gens típics ni produeixen proteïnes.
Entre els lncARNS identificats com a alterats a les regions cerebrals afectades en la síndrome de Rett es troba un que regula la funció del receptor GABA. Esta ultima molecula (cuyas sigles es refereixen a l’àcid gamma- aminobutírico) és un neurotransmisor clau en el sistema nervioso cerebral de tots els vertebrats i la seva alteració podrien explicar els defectes de comunicació neurona-neurona a les nenes afectades per la síndrome de Rett. Aquests descobriments, a més d’augmentar el coneixement sobre les causes d’aquesta malaltia, podria tenir conseqüències futures per al tractament d’aquests pacients ja que teràpies que tuvieran com diana els lncARNs o el receptor GABA mereix ser estudiades en assaigs preclínics.
L’estudi ha estat recolzat pel Departament de Salut de la Generalitat de Catalunya, la Institució Catalana d’Estudis Avançats (ICREA), el Ministerio
de Sanidad y Consumo (E-RARE), los Proyecto Europeos DISCHROM y EPINORC, la Fondation Lejeune (Francia) i l’Associació Catalana del Síndrome de Rett.
Article: Desregulació del transcriptoma llarg d’ARN no codificant en un Rett model de ratolí de síndrome. Petazzi P, Sandoval J, Szczesna K, Jorge OC, Roa L,
Sayols S, Gomez A, Huertas D, Esteller M. RNA Biology, 10(7), 2013.

La síndrome afecta gairebé exclusivament a nenes i dones. Això es deu al fet que està principalment causat per una mutació esporàdica en el gen MECP2 del cromosoma X. La funció d’aquest gen és regular el funcionament d’altres gens, per la qual cosa juga un paper fonamental en el desenvolupament cerebral.
A Espanya, unes 2.700 nenes pateixen l’anomenada Síndrome de Rett, una malaltia neurològica que afecta la mobilitat i que causa un autisme sever.
La síndrome de Rett és un trastorn neurològic infantil de base genètica caracteritzat per una evolució normal inicial seguida de la pèrdua de l’ús voluntari de les mans, un creixement retardat del cervell i del cap, dificultats per caminar, convulsions i retard mental.
Els nens que pateixen la síndrome de Rett generalment comencen a perdre la capacitat de parlar, fer contacte visual i comunicar-se d’altres maneres. Poden perdre l’interès en les altres persones, joguines i entorn. Alguns nens pateixen els canvis, com la pèrdua sobtada de la parla, ràpidament
Alteracions en altres gens com el CDLK5 i el FOXG1 també produeixen síndrome de Rett, encara que els símptomes i l’afectació poden ser molt diferents.
Aquesta síndrome es diagnostica observant signes i símptomes durant el creixement inicial i el desenvolupament de la nena i realitzant avaluacions periòdiques del seu estat físic i neurològic.
Hi ha nombrosos problemes crònics de salut i comportament associats amb RTT per als quals seria raonable esperar dolor o malestar (p. ex., escoliosi, restrenyiment i problemes gastrointestinals relacionats, comportament auto agressiu)
Existeix també una prova genètica per confirmar el diagnòstic clínic d’aquest trastorn. La prova involucra buscar la mutació de tipus MECP2 en el cromosoma X del nen o nena i pot identificar fins a un 80% dels casos.
La síndrome de Rett és una malaltia de les anomenades rares, la qual cosa significa que afecta menys de cinc casos per cada 10.000 persones. Rett és un trastorn greu del neurodesenvolupament d’origen genètic que afecta de manera gairebé exclusiva al sexe femení i que porta a una discapacitat greu. També afecta gairebé tots els aspectes de la vida d’una persona ja que impedeix la seva capacitat per parlar, caminar, menjar i fins i tot respirar de forma normal.
Malgrat les dificultats que impliquen els símptomes, la majoria dels individus que pateixen de la Síndrome de Rett continúen vivint bé fins a l’edat adulta o major.
El 1966, un neuròleg austríac anomenat Andreas Rett va descriure per primera vegada més de 20 joves pacients de sexe femení que compartien característiques similars, a partir de l’observació d’idèntics moviments estereotípics de les mans.
El diagnòstic del SR es basa en criteris clínics, però els avenços en la biologia molecular i en la genètica en particular han obert el ventall de possibilitats diagnòstiques a les diferents formes clíniques que abans quedaven sense classificar, alhora que l’anàlisi molecular permet confirmar el criteri clínic i aportar informació quant al pronòstic del pacient.